Docking
Introduction
Molecular docking is a process to couple proteins with their ligands; for this project, molecular docking was performed in order to support the decision of using the proteins that were selected as part of the biosynthetic pathway of the piperamide.
From all the enzymes used in the pathway, only 8 require a docking step for different reasons. These reasons can include the enzyme not being isolated, and though previous annotations suggest its role in the reaction, molecular docking can provide better support for selection and activity. Another reason is when the protein is known, but we are using a molecule that is not the original ligand, and molecular docking helps elucidate if the protein can perform the reaction with a similar structure.
The enzymes that require molecular docking and the genes that code for these enzymes are: Pn8.2617, Pn2.84, Pn1.1317, Pn3.4770, Pn7.1626, Pn16.1237, Pn16.1198, Pn4.3222, Pn2.2377, and 2cwh.
For each of these genes, a differential exon usage analysis was performed between four different Piper nigrumtissues: fruit at 20 days stage, fruit at 40 days stage, leaf, and panicle. This analysis helped visualize the expression level for each exon of the genes, allowing us to select the exons of the gene to include in the modeling and following steps.
This analysis was performed using the Bioconductor library "DEXSeq" and the RNA-seq reads were mapped to the reference genome of Piper nigrum using the STAR ultrafast universal RNA-seq aligner on the Galaxy project platform.
Below is a summary of the results obtained, with a couple of interesting enzymes exemplified to showcase the methodology used.
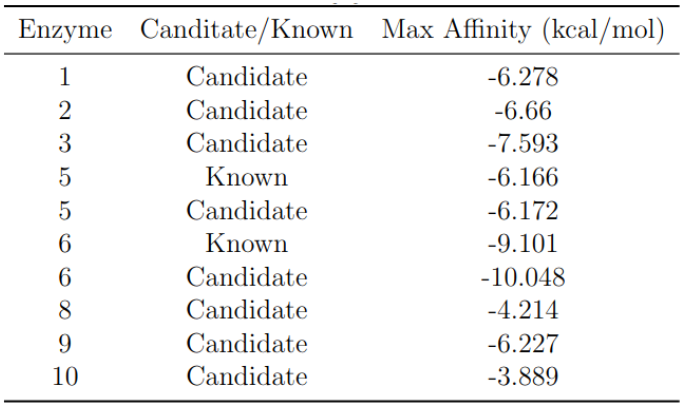
Figure 1: Overview of the molecular docking process.
Methodology
The genes selected for the docking methodology were chosen by the differential expression analysis, which proposed their viability to perform the steps required in the designed biosynthetic pathway. Structure modeling was carried out using a homology-based methodology such as “SWISS-MODEL” or AlphaFold2, utilizing the ColabFold resource.
The homology-based methodology is template-based, where the template was found using HHblits, a protein sequence searching algorithm that uses HMM-HMM alignment. The QMEANDisCo value is a composite score for single-model quality estimation, which is a function of underlying single-model scores employing statistical potentials of mean force and distance constraints based on the known structure of homologous proteins. QMEANDisCo is calculated for every residue of the protein, and the QMEANDisCo Global value is the average of these per-residue scores, representing the global overall quality of the model.
Before docking was performed, the protein and the ligand were energy minimized. Protein minimization was achieved using UCSF ChimeraX software and the steepest descent algorithm for 1000 steps with a step size of 0.02 Å; the force field AMBER ff14SB was used for standard residues, while non-standard residues were handled with the semi-empirical AM1-BCC model, which uses an additive bond charge corrections method. The ligand was minimized using the Python library “rdkit” and the MMFF94 force field.

Figure 2: Workflow of the docking methodology.
The preparation of the molecules for docking was performed with AutoDockTools, and the docking was done using Autodock Vina version 1.2.3. The grid box for docking was selected based on conserved domains found using the NCBI Conserved Domain Search interface or the CASTp3 methodology, which identifies protein pockets on the surface. CASTp3 uses computational geometry to find these pockets, mainly using Delaunay triangulation and alpha shape.
For enzymes requiring a coenzyme, a sequential docking workflow was used, as described by Vass et al. (2012). In this workflow, the coenzyme was docked first, followed by the ligand in a second step, after merging the best docking pose of the coenzyme with the protein.
Results
The best binding affinity for each enzyme is shown in the following table. The next figure shows the best nine binding affinities for each of the performed dockings.
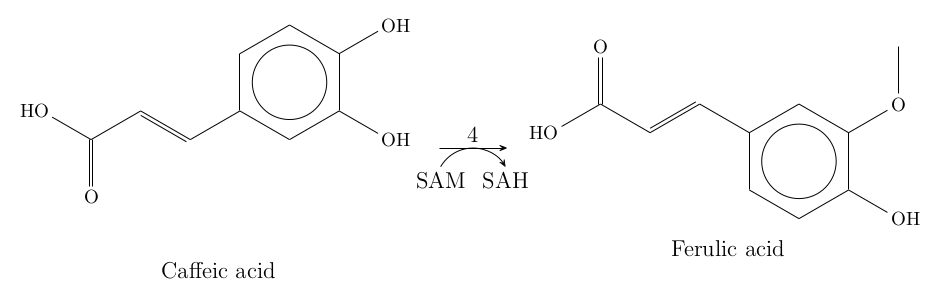
Figure 3: Best binding affinities obtained from docking simulations.
Example: Pn1.1317 Docking
The fourth reaction in the biosynthetic pathway is catalyzed by the protein caffeic acid 3-O-methyltransferase, which requires the coenzyme SAM. The docking workflow first included the minimization of the protein and the ligand, followed by docking SAM and caffeic acid. The result showed that both SAM and caffeic acid bind to the active site of Pn1.1317 with good affinities. However, the interaction of caffeic acid occurred primarily on the propanoid tail rather than the 4-hydroxy-3-methoxyphenyl region.
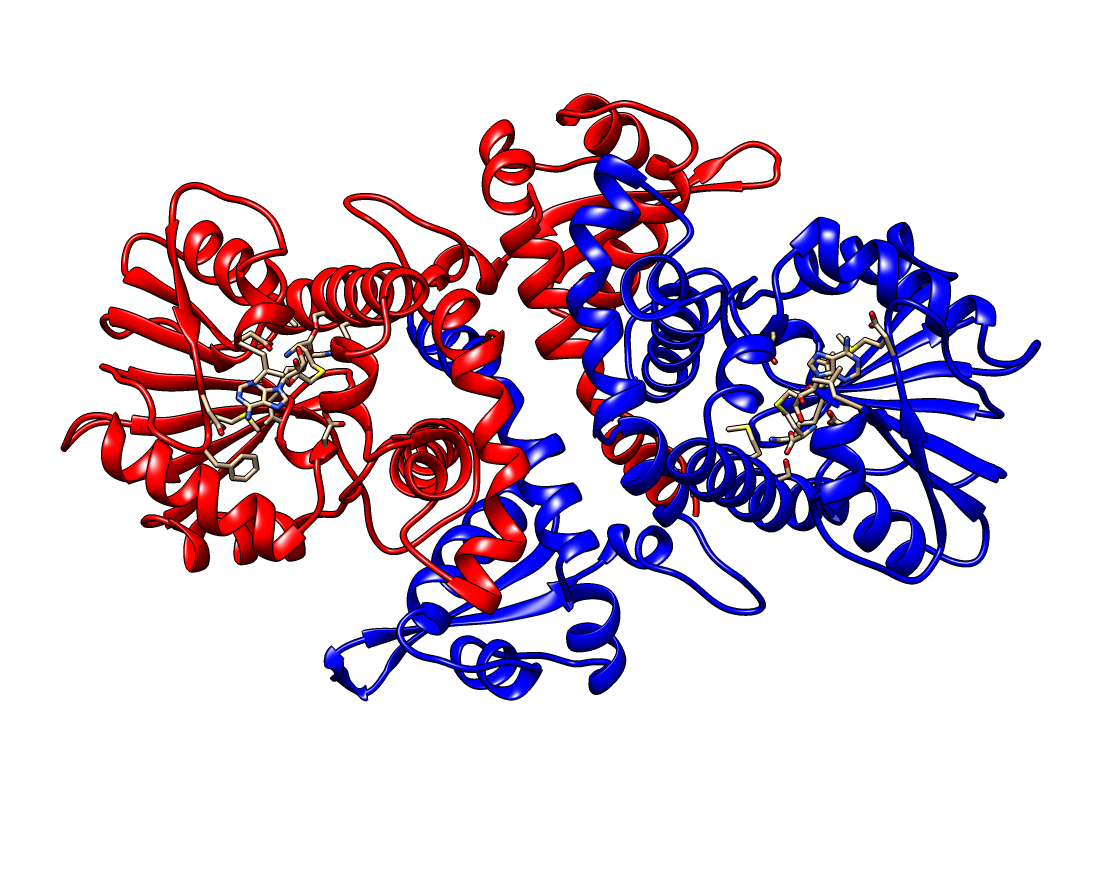
Figure 4: Docking results of SAM and caffeic acid with Pn1.1317.
Similarly, docking was performed for other enzymes, and the results are summarized below. For some enzymes, like Pn3.4770, the affinity results showed better performance for the selected docking compared to previously known enzymes, which makes it promising for further in vitro exploration.
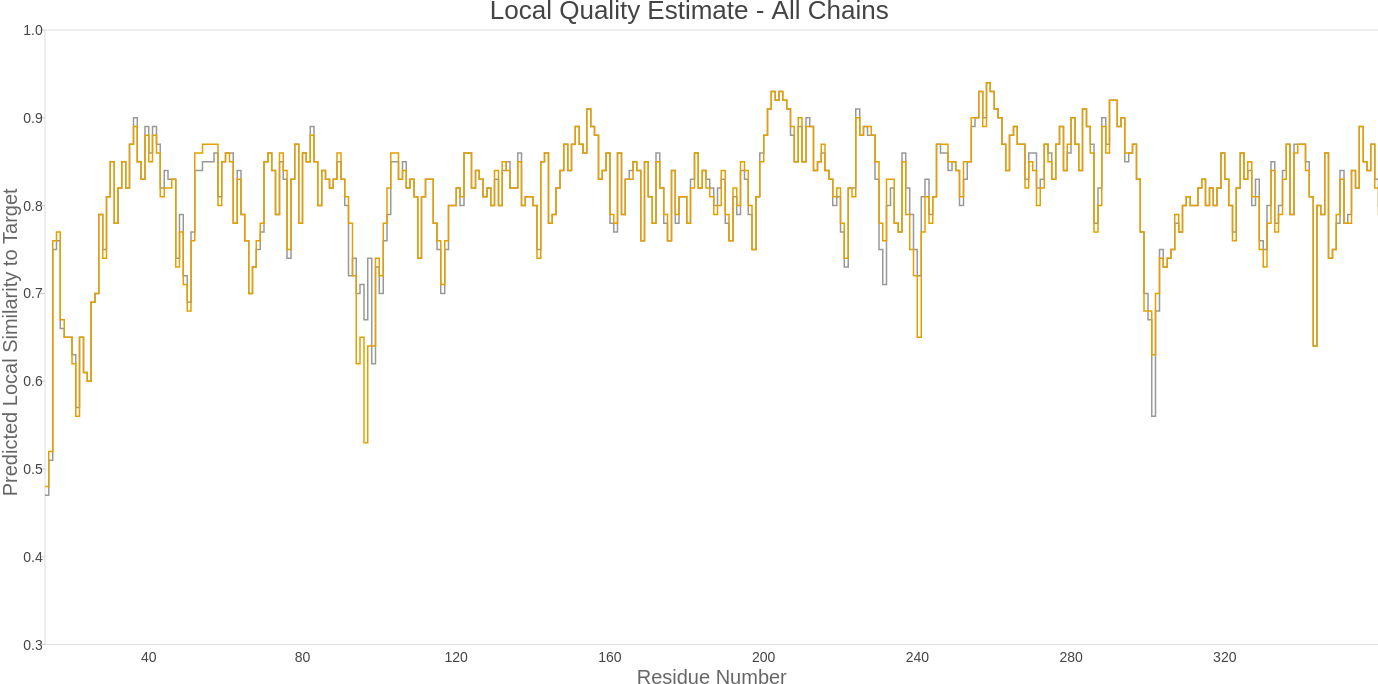
Figure 5: Docking results of ferulic acid with Pn3.4770.
The docking results help confirm the viability of using the selected enzymes for the steps of the piperamide biosynthetic pathway. Further analysis is required to test these enzymes in vitro and to optimize the reaction conditions.
Pn1.1317
The fourth reaction of the pathway is catalyzed by the protein caffeic acid 3-O-methyltransferase, which is also part of the phenylpropanoid biosynthesis pathway. The gene Pn1.1317 was selected as the best candidate for enzyme 4 based on the fact that it obtained the highest score (651.5) over the threshold (499.33) for KEGG Orthology ID K13066, with an E-value = 2.9 × 10⁻¹⁹⁶. This annotation was made with KofamKOALA. The exon usage analysis, however, did not yield relevant results.
The gene contains four exons that were considered when modeling the three-dimensional structure of the protein. The structure modeling was performed using a homology-based methodology “SWISS-MODEL”. The selected template was an O-methyltransferase from Fragaria ananassa with PDB ID: 6I71, which has a 68.39% sequence identity to our query protein Pn1.1317, making it an appropriate template. The template was found using HHblits. The model is shown in the next figure in a dimeric state, as is the template. It has a GMQE value of 0.84 and a QMEANDisCo Global value of 0.81 ± 0.05.
The enzyme requires a coenzyme SAM (S-adenosylmethionine) for its correct catalytic activity. This molecule is demethylated to SAH (S-adenosyl-L-homocysteine). The crystal structure of the template contained SAH bound to it, and the binding site for SAH was conserved in Pn1.1317. The model generated also included this molecule, and the interactions between this ligand and the protein can be seen in the next figure. The 3D conformer structures for SAM and caffeic acid were obtained from PubChem with CIDs: 34755 and 689043.
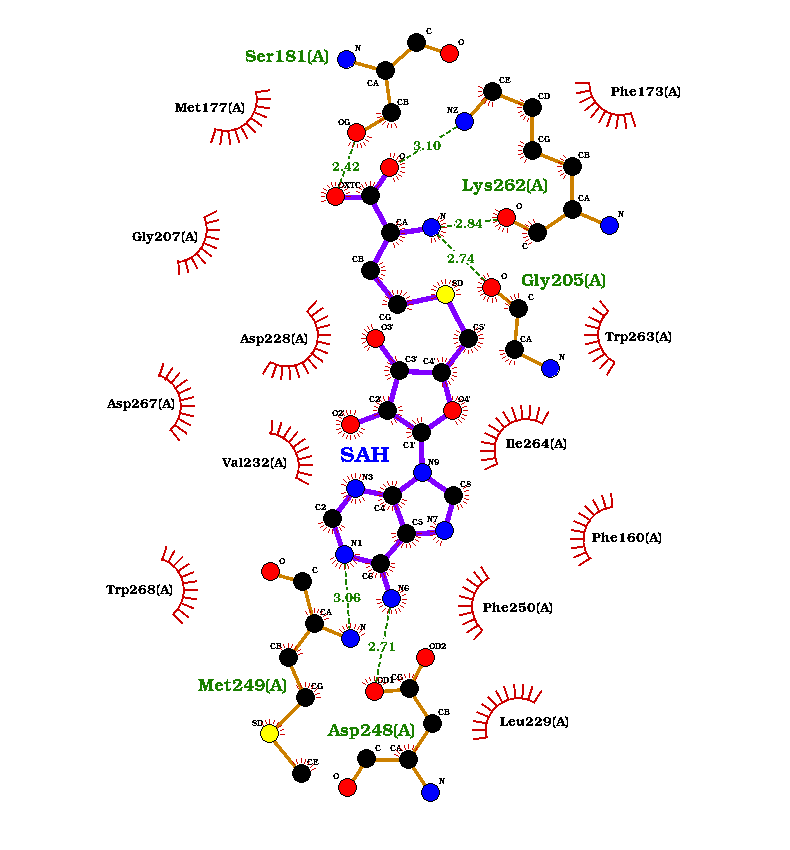
Figure 6: Interaction of SAM and caffeic acid with Pn1.1317.
Before docking was performed, the protein and the ligand were energy minimized. The grid box for the SAM docking was selected based on the conserved binding site for SAH, as found through homology modeling. To identify the binding site for caffeic acid, a blastp search against the PDB database was used to obtain similar protein sequences to Pn1.1317. Eleven sequences with identities higher than 50% were selected. These sequences were aligned using Muscle, and based on the MSA (Multiple Sequence Alignment), a search for the best amino acid substitution model was carried out using MEGA 11. The selected model was LG+G (lnL: -4126.121), a decision supported by the Bayesian Information Criterion (BIC: 8436.596) and Akaike’s Information Criterion (8296.475).
With the selected model and MSA, a Maximum Likelihood (ML) tree was constructed using MEGA 11, and branch support values were obtained from 100 Bootstrap pseudoreplicates. Using this ML tree as a starting point, a Bayesian Inference tree (BI) was built using MrBayes. Two runs of four chains (one cold chain and three heated chains) were conducted for one million generations and sampled every one thousand generations with a burn-in of 25%. The amino acid substitution model was selected by sampling all of the fixed-rate models implemented in the program, with each model contributing in proportion to its posterior probability.
This analysis was performed using the CIPRES Science Gateway portal. The results were analyzed with Tracer, which showed that the effective sample size for all parameters was higher than 200: the log likelihood of the cold chain (LnL: 1721), the log likelihood of the prior (LnPr: 1668), and the total tree length (TL: 1658).
Based on this analysis, the sequence with PDB ID: 1KYW, a caffeic acid/5-hydroxyferulic acid 3/5-O-methyltransferase from Medicago sativa, was selected to define the binding site for caffeic acid. This sequence is part of the sister group of Pn1.1317 and had the most significant alignment E-value (2 × 10⁻¹⁸⁰).
Figure 7: Binding site analysis for caffeic acid in Pn1.1317.
Pn3.4770
The fifth reaction of the pathway is catalyzed by a protein similar to CYP719A37. The gene Pn3.4770 was selected as a candidate for enzyme 5 based on its 100% identity to CYP719A37 (E-value ≊ 0) with accession number: QQS74306. The protein CYP719A37 has a length of 511 amino acid residues, but the gene Pn3.4770 codes for 578 amino acids. This indicates a possible alternative splicing process affecting the expression of this gene. Blast results and the exon usage analysis revealed that from the three exons present in gene Pn3.4770, only the first two exons were expressed.
Only the first two exons of the gene were considered when modeling the three-dimensional structure of the protein. The structure modeling was performed using an artificial intelligence-based methodology. The algorithm used for the 3D structure prediction of the protein was AlphaFold2 via the ColabFold resource. ColabFold is a collaborative platform that allows users to perform three-dimensional structure predictions using AlphaFold2, AlphaFold2 multimer, MMseqs2, and HHsearch, all without the need for a local supercomputer. The Local Distance Difference Test (lDDT) score for the model is displayed in the next figure, which assesses the local quality of the model.
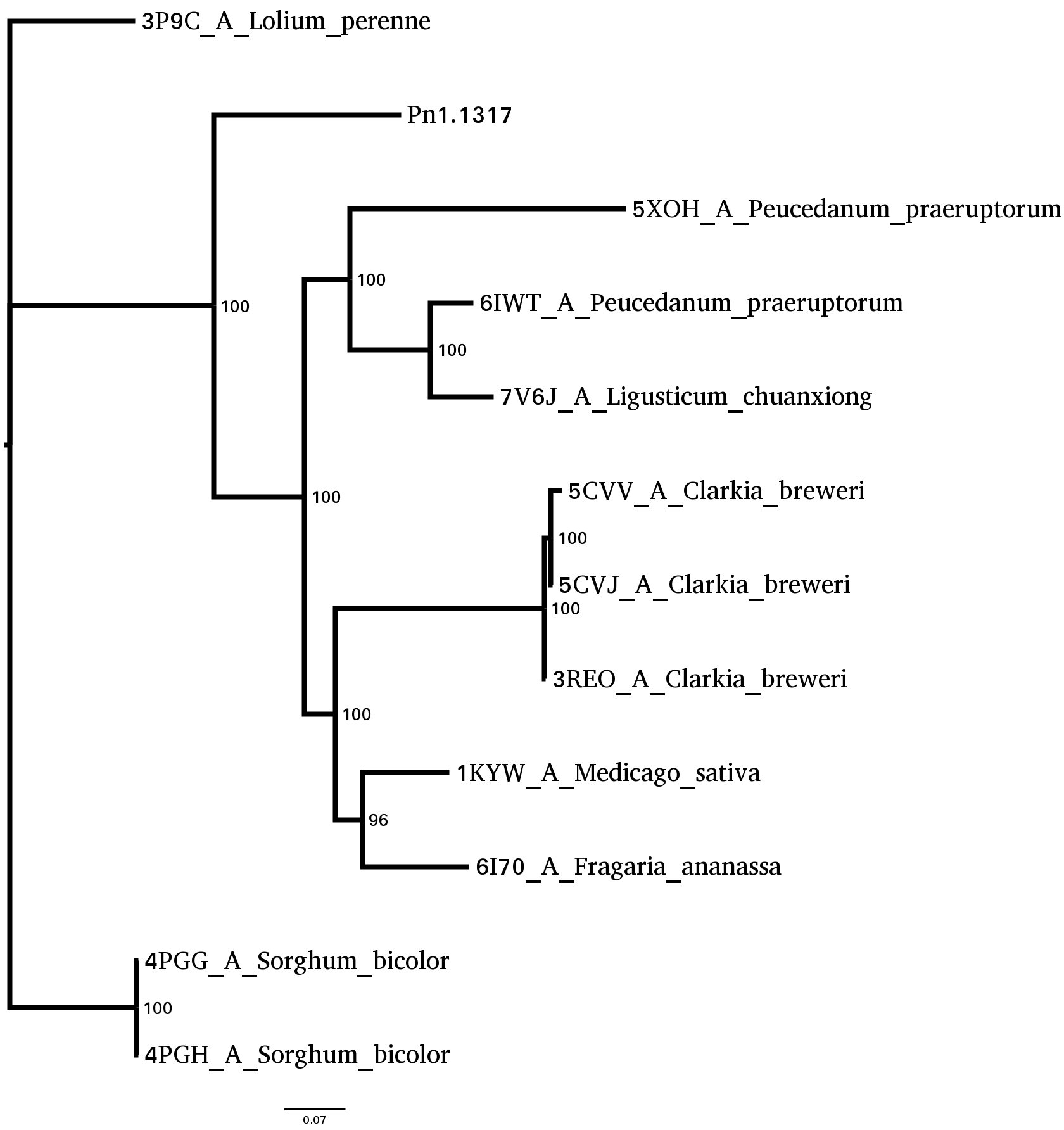
Figure 8: Local Distance Difference Test (lDDT) score for Pn3.4770.
The enzyme requires a heme molecule as a coenzyme for its correct catalytic activity. In this case, protoporphyrin IX containing Fe was selected based on the heme group present in the template for a SWISS-MODEL structure prediction. The template used was the CYP-450 17A1 protein from Danio rerio with PDB ID: 6b82, though this model was not chosen due to the low sequence identity with Pn3.4770 (24.62%). The ligand 3D conformer structure was obtained from PubChem with CID: 445858.
The sequence used in this methodology (CYP719A37) has been isolated by Schnabel et al. (2021b), and its activity has been tested with various substrates. Its original substrate is feruperic acid, which has a two-carbon extension compared to ferulic acid. When ferulic acid was used as a substrate, the expected product (3,4-methylenedioxy cinnamic acid) could not be detected. This suggests that the enzyme may not catalyze the reaction, but docking was still performed to compare the results of this enzyme with the proposed enzyme that could perform the reaction.
The first molecule docked was protoporphyrin IX with Fe. As a result of molecular docking using Vina, nine different conformations of the molecule in the protein were found. The docked positions of protoporphyrin IX with Fe in the Pn3.4770 structure interacted with several amino acid residues of the protein. Some of these amino acids were found in the conserved domain for the heme binding site of the enzyme, including Arg 119, Leu 156, Leu 206, Val 305, Leu 308, and Gly 309.
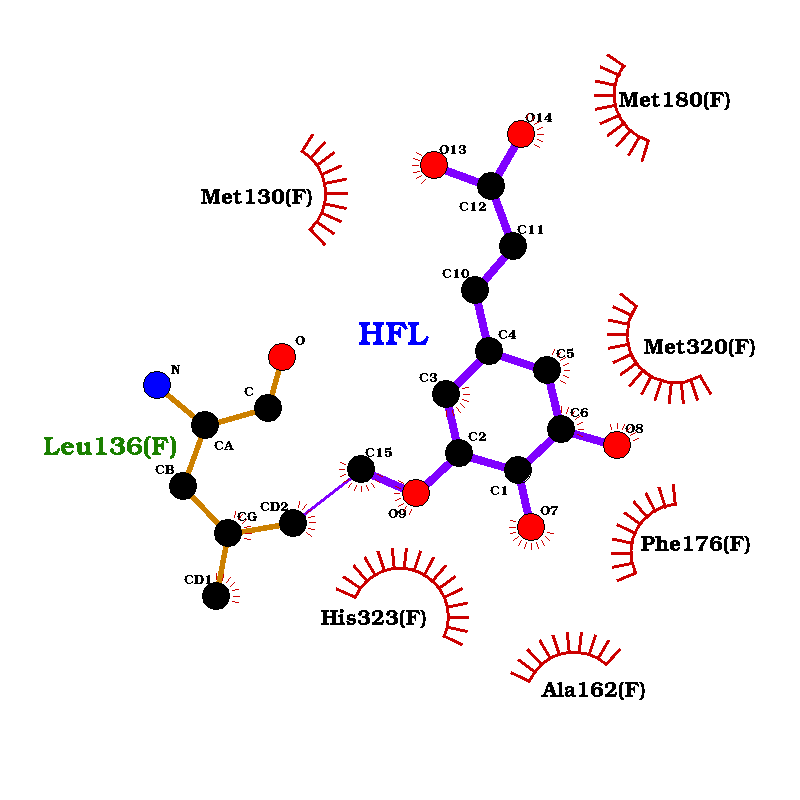
Figure 9: Docking of protoporphyrin IX with Fe in Pn3.4770.
With the Pn3.4770 protein and the best docked pose of protoporphyrin IX with Fe as a complex, molecular docking using Vina was carried out for ferulic acid, resulting in nine different conformations. The docked positions of ferulic acid in the receptor structure interacted with several amino acid residues of the protein, such as Leu 307, Leu 308, and Val 379, which are part of the conserved domain for the active site of the enzyme.
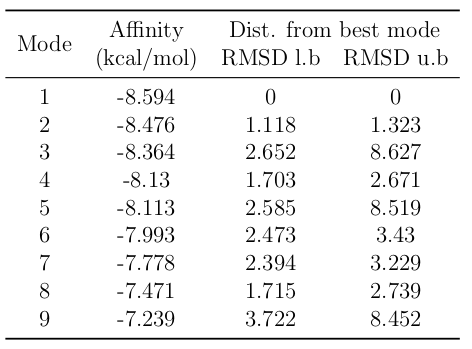
Figure 10: Docking of ferulic acid in Pn3.4770.
The results show that interactions between ferulic acid and the Fe atom of protoporphyrin IX occur primarily on the propanoid tail rather than the 4-hydroxy-3-methoxyphenyl region, suggesting that this enzyme may have limited activity with ferulic acid as a substrate.
References
- Altschul, S. F., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25(17), 3389–3402.
- Altschul, S. F., et al. (2005). Protein database searches using compositionally adjusted substitution matrices. The FEBS Journal, 272(20), 5101–5109.
- Anders, S., et al. (2012). Detecting differential usage of exons from RNA-seq data. Nature Precedings, 1–1.
- Aramaki, T., et al. (2020). KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics, 36(7), 2251–2252.
- Bertoni, M., et al. (2017). Modeling protein quaternary structure of homo-and hetero-oligomers beyond binary interactions by homology. Scientific Reports, 7(1), 1–15.
- Community, T. G. (2022). The Galaxy platform for accessible, reproducible, and collaborative biomedical analyses: 2022 update. Nucleic Acids Research, 50(W1), W345.
- Dobin, A., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15–21.
- Eberhardt, J., et al. (2021). AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. Journal of Chemical Information and Modeling, 61(8), 3891–3898.
- Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797.
- Goddard, T. D., et al. (2018). UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Science, 27(1), 14–25.
- Waterhouse, A., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303.
- Zubieta, C., et al. (2002). Structural basis for the modulation of lignin monomer methylation by caffeic acid/5-hydroxyferulic acid 3/5-O-methyltransferase. The Plant Cell, 14(6), 1265–1277.